Week 4, Session 1 — RNA-seq with DESeq2 / edgeR
Course 4 — #courses
2. Approach
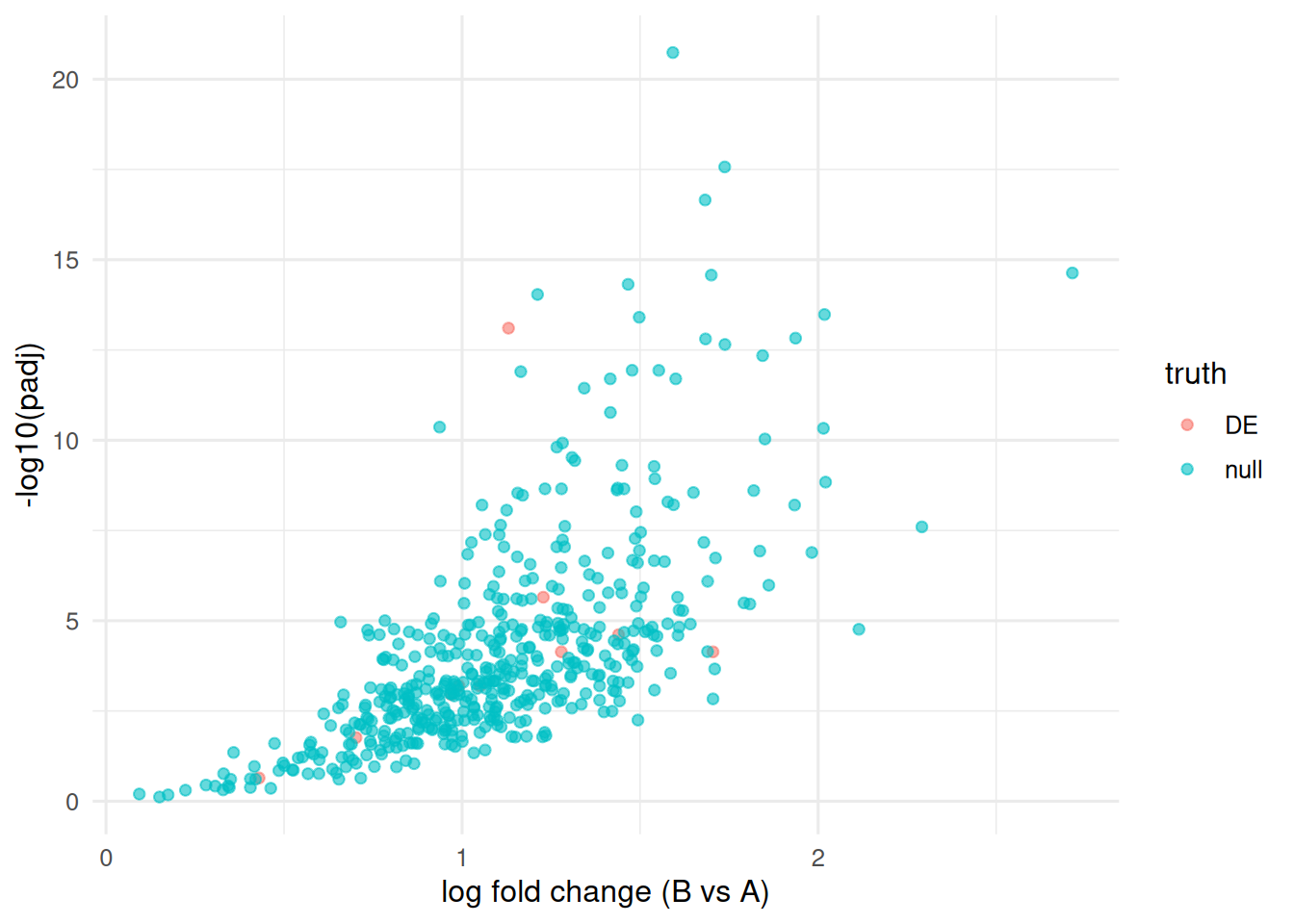
Simulate 500 genes × 10 samples (5 per group). Ten genes are truly differentially expressed.
n_gene <- 500; n_samp <- 10
group <- rep(c("A", "B"), each = 5)
mu <- 2 * runif(n_gene, 1, 10) # baseline mean per gene
lfc <- c(rep(log(3), 10), rep(0, n_gene - 10))
counts <- sapply(seq_len(n_samp), function(j) {
rate <- mu * exp(ifelse(group[j] == "B", lfc, 0))
rnbinom(n_gene, mu = rate, size = 5)
})
rownames(counts) <- paste0("g", seq_len(n_gene))
colnames(counts) <- paste0("s", seq_len(n_samp))
tibble(mean = rowMeans(counts), var = apply(counts, 1, var)) |>
ggplot(aes(mean, var)) + geom_point(alpha = 0.3) +
scale_x_log10() + scale_y_log10() +
geom_abline(slope = 1, intercept = 0, colour = "grey50") +
labs(title = "mean–variance (log–log)")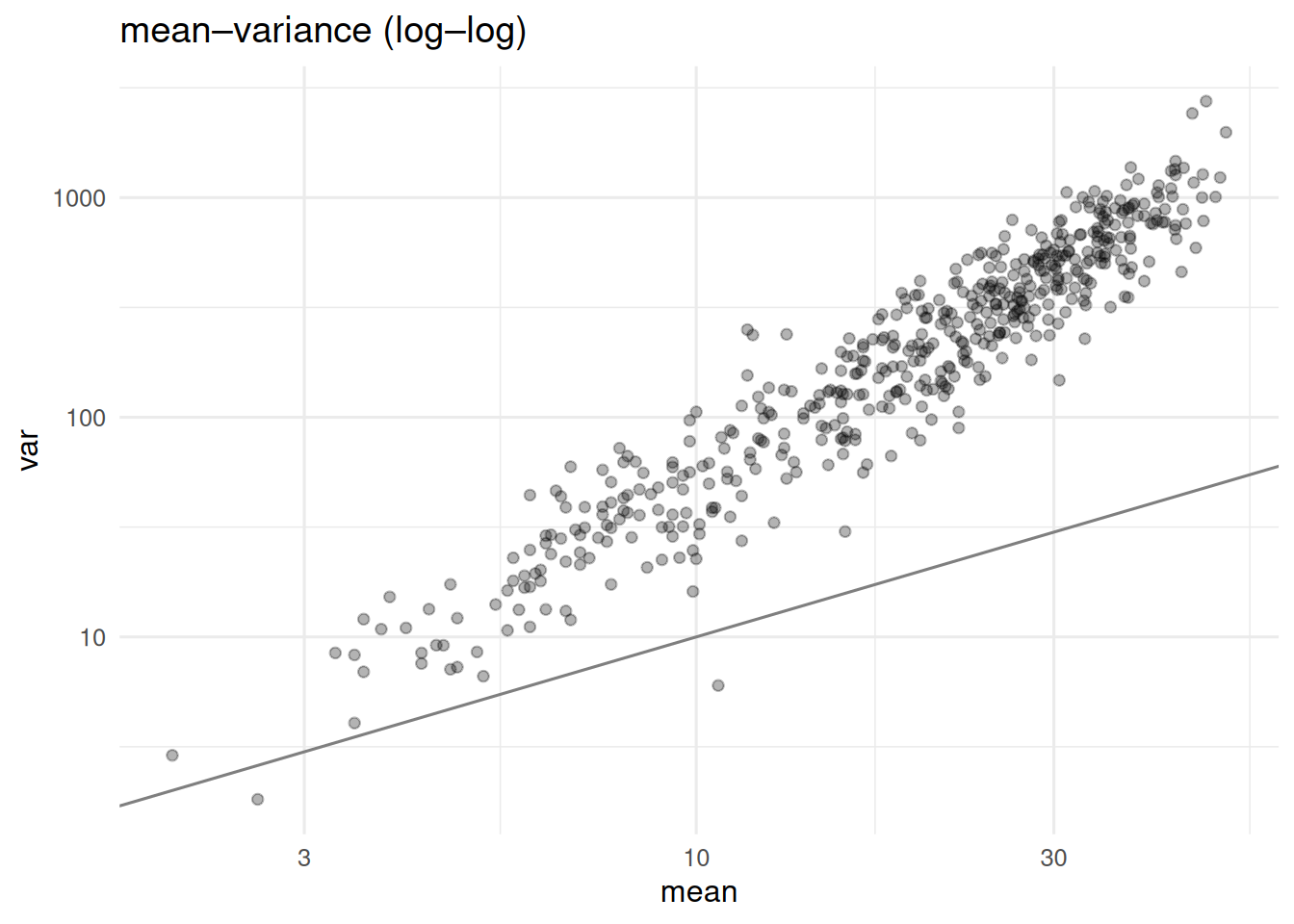
4. Check
Volcano plot with the truly DE genes flagged.