![]()
Overview
phenoscapR provides tools for reading, processing, analysing, and visualising single-cell spatial biology data from multiplexed imaging platforms.
A typical workflow involves:
-
Reading cell segmentation data with
read_spatial() -
Quality control with
qc_filter() -
Normalisation with
normalise_markers() -
Phenotyping with
phenotype_cells() -
Spatial analysis with
nearest_neighbours(),cell_density(),interaction_matrix(), andspatial_clusters() -
Visualisation with
plot_cell_map(),plot_density(),plot_heatmap(), andplot_interactions()
Creating Example Data
Since real cell segmentation files can be large, we will create a small simulated data set for illustration.
library(phenoscapR)
set.seed(42)
n_cells <- 500
# Simulate two cell populations in different spatial regions
dt <- data.table::data.table(
`Cell ID` = seq_len(n_cells),
`Cell X Position` = c(rnorm(250, 200, 50), rnorm(250, 600, 50)),
`Cell Y Position` = c(rnorm(250, 300, 50), rnorm(250, 300, 50)),
`Cell Area (px)` = rlnorm(n_cells, log(100), 0.3),
CD3 = c(rnorm(250, 800, 150), rnorm(250, 200, 100)),
CD8 = c(rnorm(250, 200, 100), rnorm(250, 700, 120)),
DAPI = rnorm(n_cells, 1000, 200)
)
# Write to a temporary CSV
tmp <- tempfile(fileext = ".csv")
write.csv(dt, tmp, row.names = FALSE)Reading Data
cells <- read_spatial(tmp, sample_id = "example")
head(cells)
#> sample_id cell_id x y cell_area CD3 CD8 DAPI
#> <char> <int> <num> <num> <num> <num> <num> <num>
#> 1: example 1 268.5479 351.4570 200.87599 709.7926 225.0578067 1123.4673
#> 2: example 2 171.7651 345.7387 117.02725 779.6276 172.2075951 999.0918
#> 3: example 3 218.1564 299.8772 133.80590 651.9091 27.5264266 981.7487
#> 4: example 4 231.6431 306.8005 111.97350 924.7888 -0.6704944 1079.9919
#> 5: example 5 220.2134 263.9923 74.17226 680.7411 70.8191670 1117.7803
#> 6: example 6 194.6938 290.0938 83.59012 851.0697 236.5838228 996.6244Normalisation
cells <- normalise_markers(cells, method = "zscore")
summary(cells[, c("CD3", "CD8", "DAPI")])
#> CD3 CD8 DAPI
#> Min. :-1.6156 Min. :-1.93580 Min. :-2.97169
#> 1st Qu.:-0.9104 1st Qu.:-0.92825 1st Qu.:-0.63287
#> Median :-0.2684 Median :-0.05788 Median : 0.05662
#> Mean : 0.0000 Mean : 0.00000 Mean : 0.00000
#> 3rd Qu.: 0.9048 3rd Qu.: 0.90400 3rd Qu.: 0.60278
#> Max. : 2.5022 Max. : 2.39231 Max. : 2.70425Phenotyping
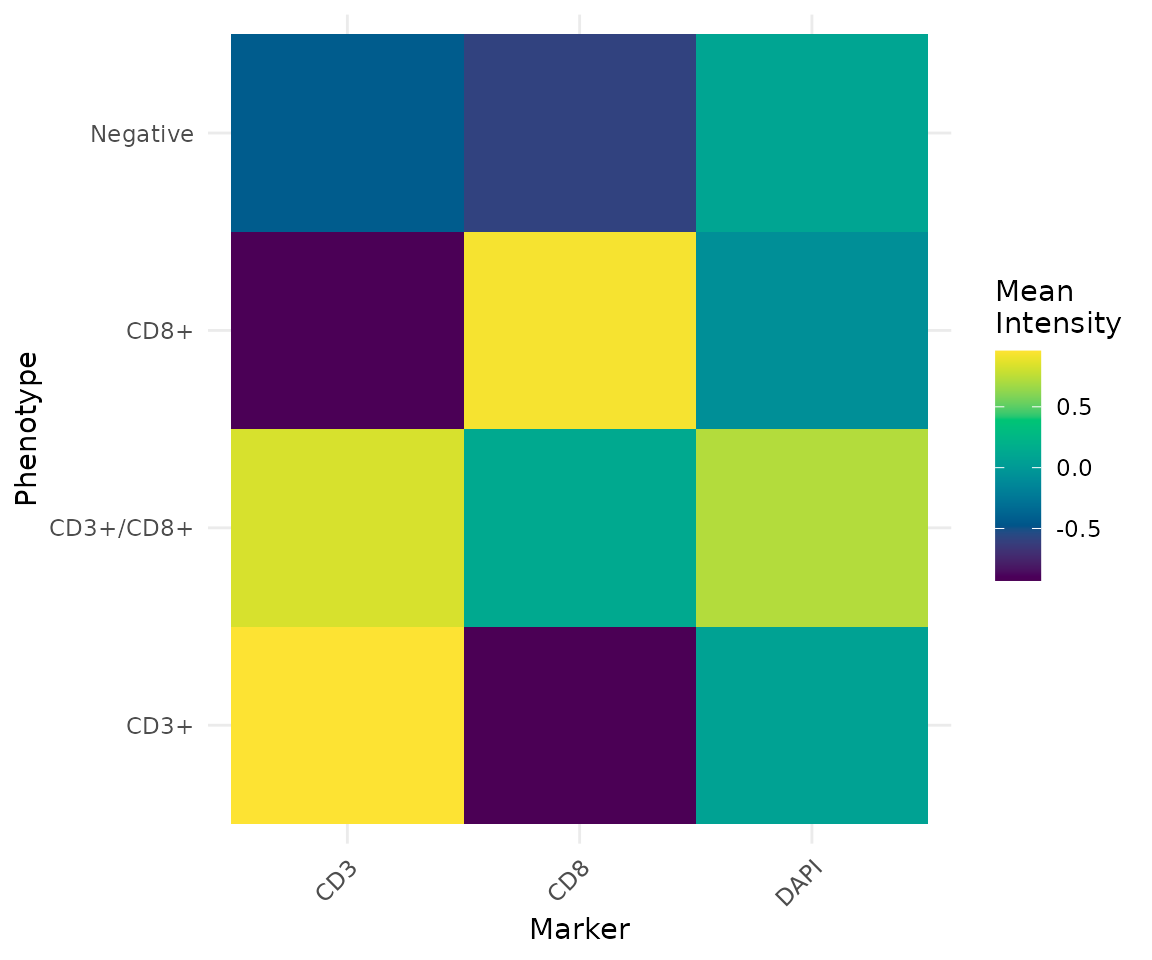
cells <- phenotype_cells(cells, thresholds = list(CD3 = 0, CD8 = 0))
summarise_phenotypes(cells)Spatial Analysis
Nearest Neighbours
cells <- nearest_neighbours(cells, k = 5)
summary(cells$nn_distance)
#> Min. 1st Qu. Median Mean 3rd Qu. Max.
#> 4.794 7.516 10.435 13.540 15.533 89.053Cell Density
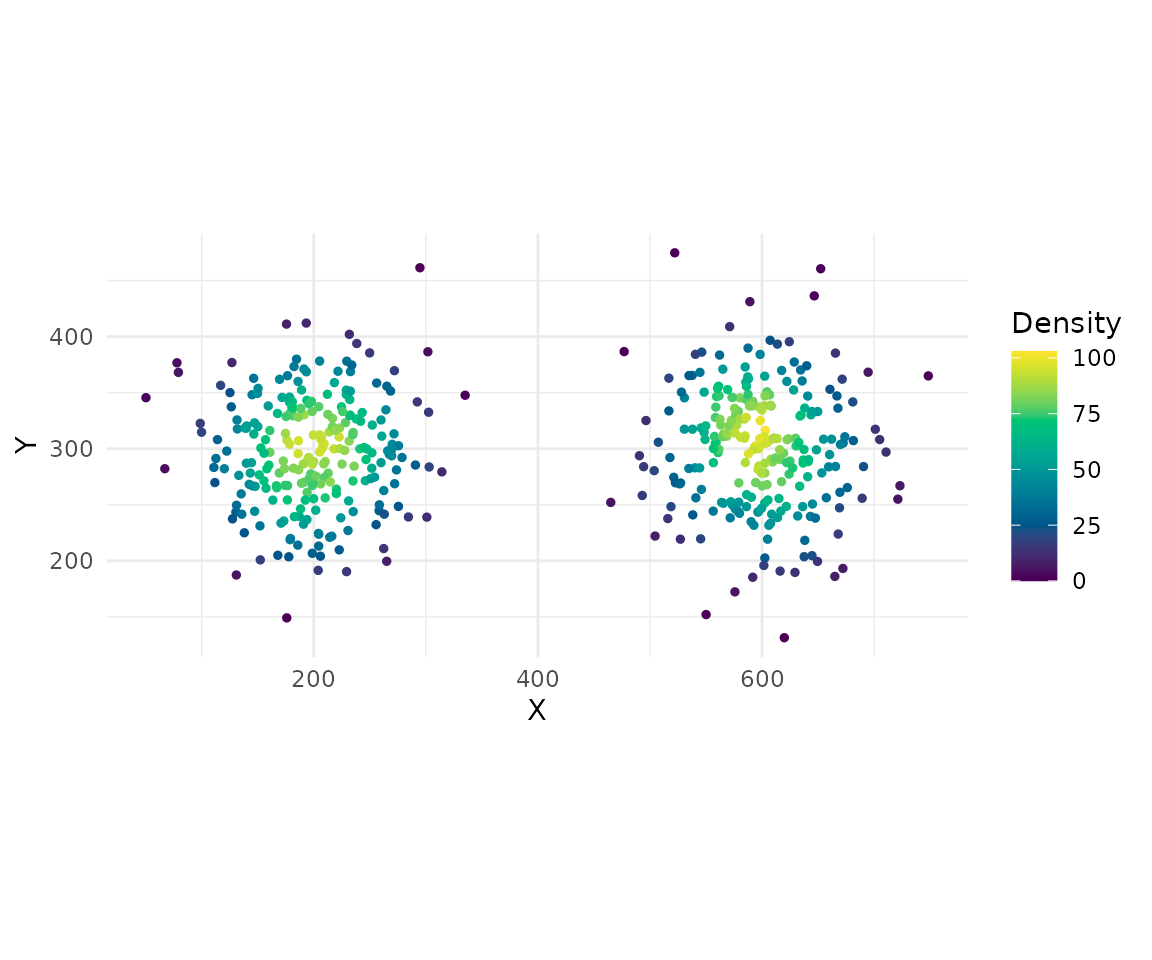
cells <- cell_density(cells, radius = 50)
summary(cells$density)
#> Min. 1st Qu. Median Mean 3rd Qu. Max.
#> 0.00 33.00 55.00 53.76 77.00 103.00Interaction Matrix
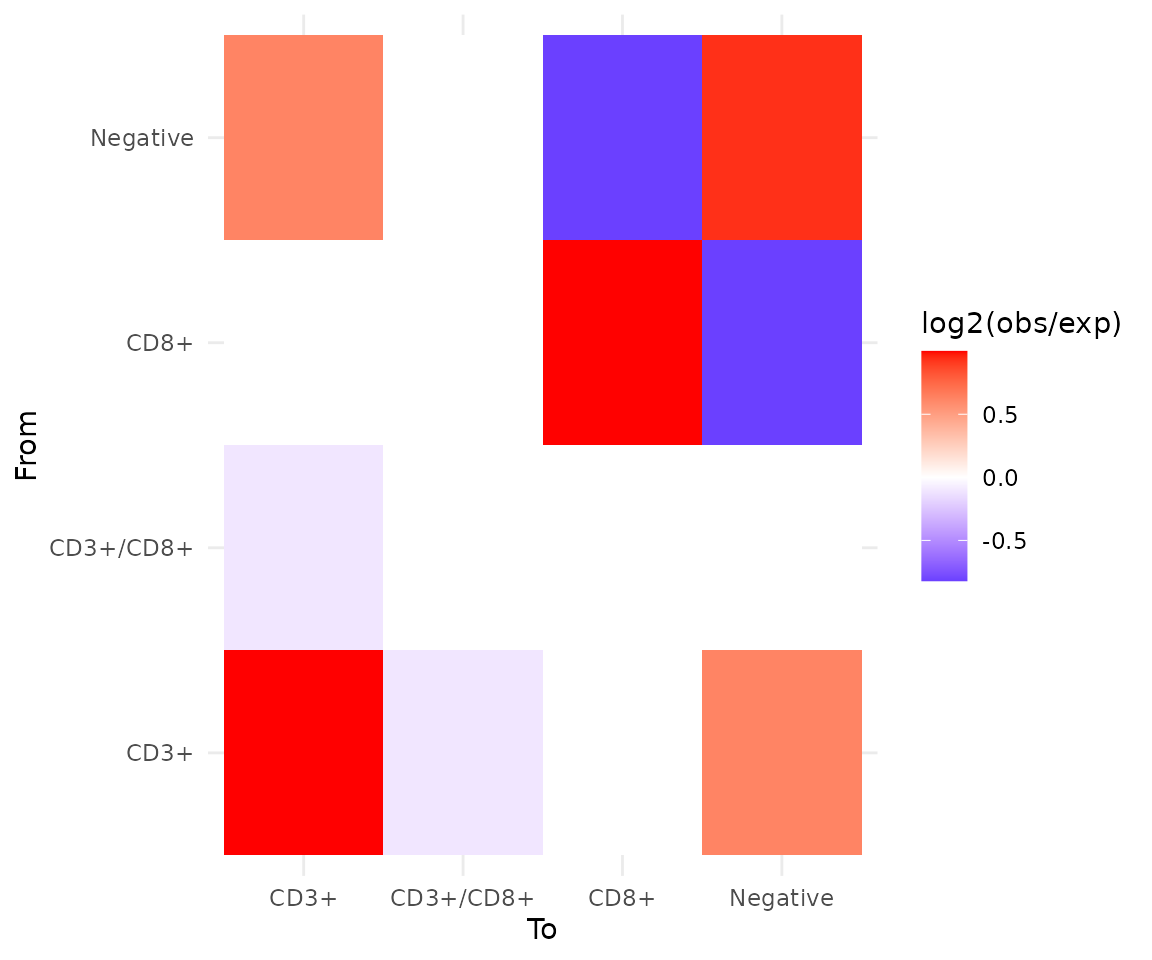
interactions <- interaction_matrix(cells, radius = 50)
interactions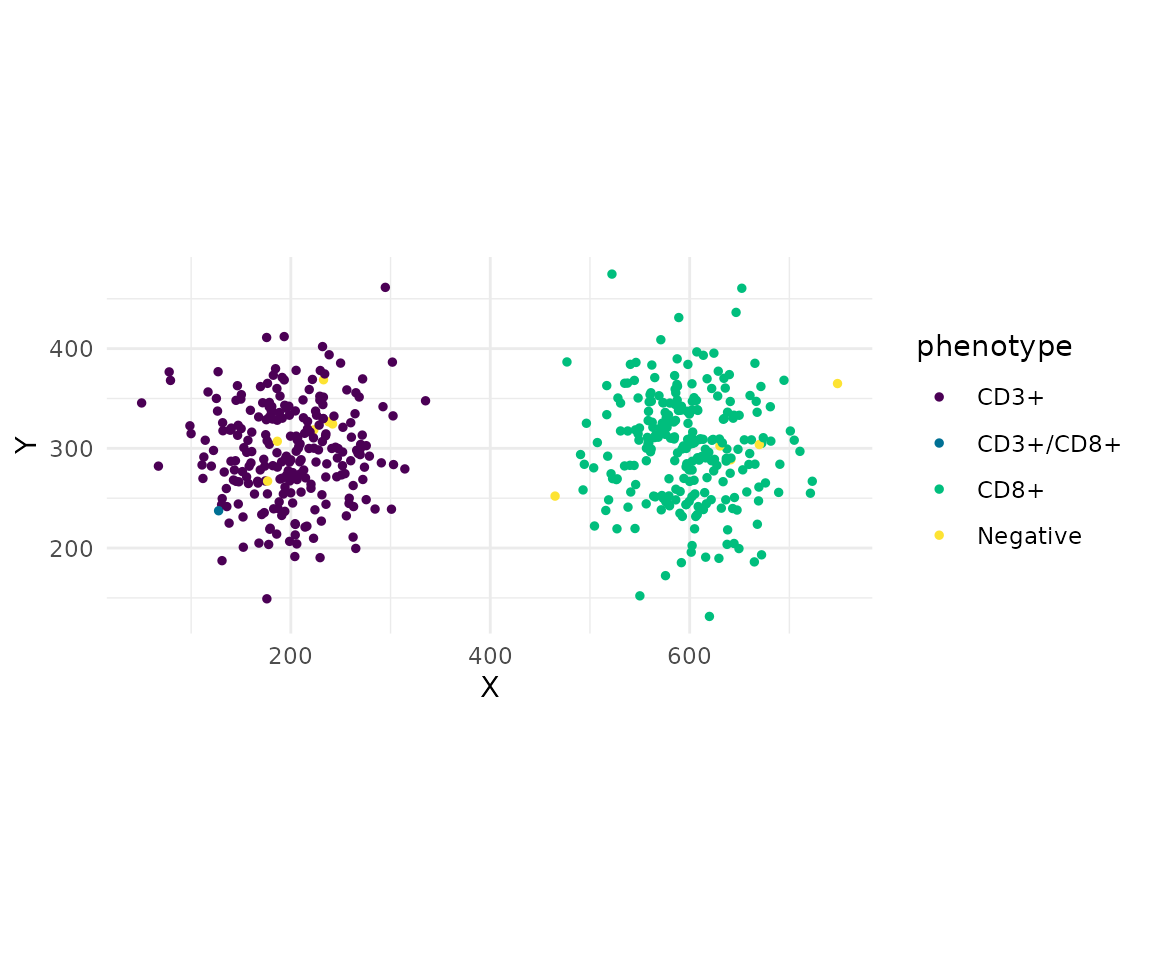
Use of LLM tools
Portions of this package were prepared with assistance from large
language model tooling for narrowly defined, non-authorial tasks:
copyediting, prose smoothing, Markdown/LaTeX formatting, scaffolding of
boilerplate files (CI configs, build scripts), code refactoring. The
tools used were Chat AI,
the LLM service of KISSKI (GWDG), and a self-hosted Mistral
Small (24B, Apache-2.0) run locally via Ollama and the ollamar R
package — local inference only, with no data sent to third parties for
the self-hosted model.
All scientific claims, methodological choices, analyses, interpretations, and conclusions are the author’s own. No LLM-generated text was incorporated without review and revision, and every reference was verified against its DOI, arXiv ID, or ISBN.